
Webinar series on
Light-Matter Interaction
This series of webinars aims at discussing exciting new concepts and developments related to light-matter interaction. They also offer an occasion to interact with early-career researchers and discuss stimulating new ideas in the field of photophysics and photochemistry – compensating somehow for the discussions one may have during a workshop.
If you are interested in joining our webinars, please contact Basile directly.
You will find below the full list of confirmed webinars:
24 September 2020 – 4.30pm (BST)
Mixed Quantum-Classical Dynamics in Cavity Quantum Electrodynamics
by Dr. Norah Hoffmann (Columbia University)
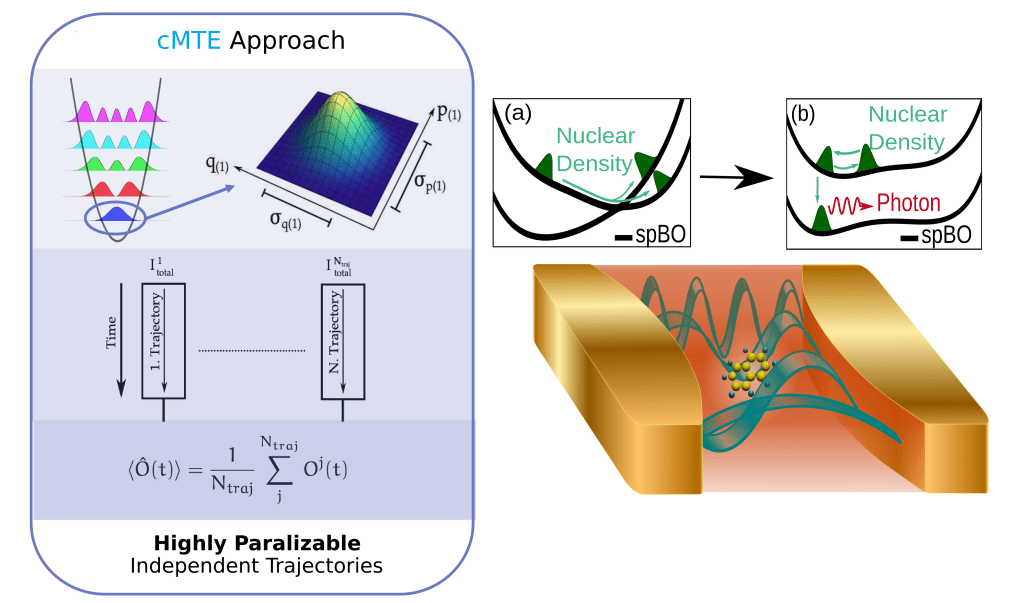
Chemical reactions under the influence of light play a paramount role in everyday life, from fundamental biological processes such as photosynthesis, vision, and DNA radiation-damage, to sustainable energy applications such as solar cells. Thus, controlling or optimizing the underlying chemical processes is of great interest. Recently, experimental developments in cavity Quantum Electrodynamics (QED) have opened up new possibilities of modifying and controlling chemical reactions by taking advantage of the strong light-matter interaction that arises when matter is confined in cavities. In such experiments the quantized nature of light becomes important and can profoundly change the chemical landscape. However, investigating these new developments from the theoretical point of view presents a challenging and especially high-dimensional problem, i.e. many molecules (electrons and nuclei) interacting with quantized light fields (many photon modes). Therefore, the theoretical simulation and prediction of realistic cavity QED experiments is by far not a trivial task and requires accurate and computationally efficient approximations.
To this end, we investigated and benchmarked extensions of mixed-quantum classical trajectory methods and time-dependent potential energy surfaces, both traditionally introduced for electron-nuclear problems, to the photonic degrees of freedom. In this talk I will discuss, how Wigner-sampling schemes for photons enable us to introduce quantized light-fields and multi-photon-mode treatments to quantum dynamics simulations in a computational efficient way, by properly accounting for the quantum statistics of the vacuum field while using classical/semi-classical trajectories to describe the time-evolution. Additionally, I will give an overview of our latest work on extending the exact-factorization approach to the photonic degrees of freedom, in order to set a starting point for the development of new mixed-quantum classical approaches within strongly coupled light-matter systems.
15 October 2020 – 2pm (BST)
Developing and applying efficient DD-vMCG method for nonadiabatic simulations
by Georgia Christopoulou (UCL)
Direct Dynamics is the branch of molecular dynamics simulations that solves the time- dependent Schrödinger equation (TDSE) by allowing the calculation of potential energy surfaces on- the-fly [1]. One of the major advantage of this method is that it is feasible to treat any system available to quantum chemistry as simply as using a modern quantum chemistry computer program. Also, since a quantum dynamics method is used to propagate the nuclei, the analysis of the influence of the quan- tum effects on reactivity is enabled. In the Direct Dynamics variational multi-configuration Gaussian wavepacket (DD-vMCG) method, a fully variational solution of the TDSE is made for the nuclei. The nuclear wavefunction is described as a superposition of Gaussian Wavepackets and the potential surfaces are provided on-the-fly [2]. During every timestep in a DD-vMCG propagation the calculated energies, gradients and hessian matrix are stored in a database. One important challenge of this method is the time needed to continually reread, sort and analyze this database which makes the calculation of a large system very expensive. To this end, the aim is to improve the existing method to be more efficient so we can treat complex chemical systems. In this talk our recent methodological updates to the DD-vMCG implementation in the Quantics sets of programs along with benchmark calculations on the allene radical cation will be discussed in detail.
[1] G. A. Worth, M. A. Robb, and B. L. Lasorne. Mol. Phys., 106:2077–2091, (2008).
[2] B. Lasorne, M.A. Robb, and G.A. Worth. PCCP, 9:3210 – 3227, (2007).

12 November 2020 – 2pm (UK Time)
Intramolecular Singlet Fission Mechanism in Donor-Acceptor Copolymers: Nonadiabatic Quantum Dynamics
by Dr. Maria Fumanal (EPFL)
Singlet Fission (SF) is a photophysical phenomenon that promises to overcome the limit of photo-conversion efficiency in organic photovoltaics by converting one singlet into two triplet excitons following S1→1TT→2T1. Since many years, SF has been reported for molecular crystals as well as isolated dimers through intermolecular and intramolecular mechanism, respectively. The latter is particularly attractive because the SF efficiency is not dependent on the crystal packing but an intrinsic property much easy to tune via molecular design. In this context, the donor-acceptor (D-A) modular strategy of copolymers has shown great potential for SF given that it can incorporate low-lying charge-transfer (CT) and multi-excitonic states in a single polymer chain [1-2]. Few D-A copolymers have remarkably succeed in displaying intramolecular SF (iSF), however the reasons behind the extraordinary efficiency found in some of these systems remains unknown. SF is an intrinsically dynamic process that can be hardly understood from the static picture considering that several (de)activation channels may compete with one another. To this end, we investigated how singlet splitting S1→1TT occurs in a prototypical iSF D-A copolymer by means of nonadiabatic quantum dynamics simulations. In this talk, I will discuss how to comprehensively build up a model Hamiltonian that serves as the basis for wave-packet propagations performed in a set of coupled diabatic potentials. This strategy allows to identify which excited states, vibrations and electronic coupling drive the ultrafast excited state decay in these systems and reveal the interplay between a direct, a mediated and a potentially delayed mechanism.
[1] Busby et al. Nat. Mater. 2015, 14, 426
[2] Blaskovits et al. Chem. Mat. 2020, 32, 6515

11 February 2021 – 2pm (UK Time)
Accelerating nonadiabatic dynamics simulations with deep learning techniques
by Dr. Julia Westermayr (University of Warwick)
Nonadiabatic molecular dynamics simulations are powerful tools to unravel the mechanisms that take place in molecular systems after light excitation, but their accurate simulation is limited by the high complexity and computational efforts involved in the underlying electronic structure calculations [1]. In this talk, I will discuss how deep learning algorithms can help to provide an analytical description of the excited states, i.e., excited-state energies, forces, couplings, and dipole moments, by exploiting the underlying physics of the data. Therefore, I will introduce the SchNarc approach [2] that allows to fit all the before-mentioned excited-state properties simultaneously. I will demonstrate the accuracy of its deep-learned potentials via UV/visible absorption spectra [3] and nonadiabatic dynamics simulations using the methylenimmonium cation as an example [2,4].
[1] J. Westermayr, P. Marquetand, Chem. Rev., in press, doi:10.1021/acs.chemrev.0c00749 (2020).
[2] J. Westermayr, M. Gastegger, P. Marquetand, J. Phys. Chem. Lett. 11(10), 3828-3834 (2020).
[3] J. Westermayr, P. Marquetand J. Chem. Phys. 153, 154112 (2020).
[4] J. Westermayr, M. Gastegger, M. Menger, S. Mai, L. González, P. Marquetand, Chem. Sci. 10, 8100-8107 (2019).

8 April 2021 – 2:30pm (UK Time)
Azobenzene photoisomerization: the role of torsion/inversion coordinates and substitution effects
by Flavia Aleotti (University of Bologna)
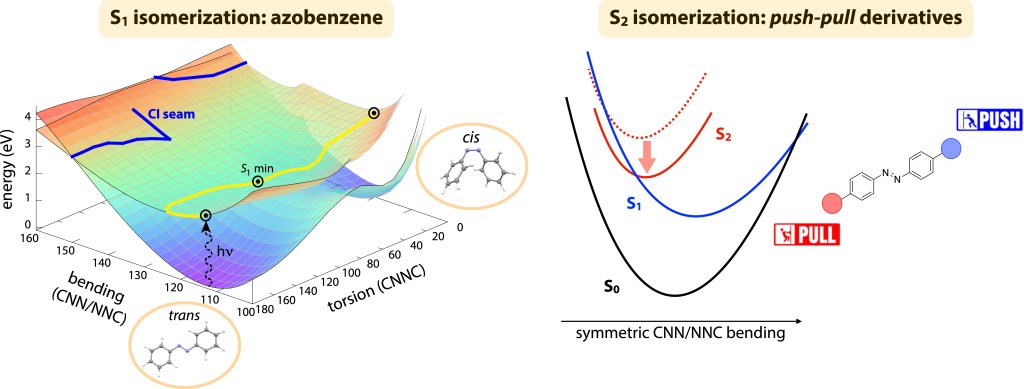
The azobenzene molecule, in which two phenyl rings are linked by a N=N unit, is a simple chromophore whose ability to undergo cis-trans photoisomerization has long been known.1 Even if the simplicity of the molecular system, together with the stability of both isomers, make this molecule a perfect candidate for applications as a molecular photoswitch, the mechanism behind its isomerization has challenged chemists and physicists for over eighty years. The experimental evidence that the quantum yield depends on the excitation wavelength suggests that different reaction mechanisms can take place starting from different excited states. Indeed, the absorbing bright state (S2, ππ*) shows a significantly lower efficiency compared to the lower nπ* state (S1) which, however, is optically dark. Historically, two main reaction paths were proposed, whose relevance in the isomerization dynamics has long been debated: rotation around the central N=N bond (torsion) and inversion at one of the two CNN angles.
We have mapped the potential energy surfaces (PESs) of the ground and first excited state of azobenzene along three coordinates describing the torsion and inversion mechanisms (i.e. CNNC dihedral and CNN/NNC angles) using state-of-the-art ab initio RASPT2 computations with an 18 electrons in 16 orbitals active space.2 Besides the energies, we have also computed other relevant quantities describing the coupling between the states, such as transition dipole moments (TDMs) and non adiabatic couplings (NACs). The adiabatic state minima and crossing points obtained with the reduced-dimensionality scan were benchmarked against fully-unconstrained optimizations at the same level of theory, and proved to be very accurate. In agreement with the most recent studies on azobenzene,3 we find that the photoisomerization mechanism from S1 is mainly torsional, but the contribution of the bending coordinate is crucial to couple the states: in particular, treating the two CNN bending angles as independent coordinates is fundamental to break the symmetry and reach the crossing region. The data that we produced allows to simulate the photoisomerization dynamics with different techniques: from simple semi-classical dynamics in the three-dimensional subspace to accurate quantum dynamics simulations, that were conducted in collaboration with the group of Professor Shaul Mukamel (University of California, Irvine) to simulate X-ray ultrafast diffraction experiments on the photoisomerization event.4The study of the photoisomerization mechanism was also extended to azobenzene push-pull derivatives, whose substituents can red-shift the bright state absorption up to the visible range, which is a key property for many biological and technological applications. We have performed semiclassical dynamics simulations (CAM-B3LYP/6-31G*) on azobenzene and two push-pullderivatives starting from the bright S2 state (ππ*).5 Our results show that the push-pull substitution is also able to increase the ππ* photoisomerization quantum yield, compared to pure azobenzene: this is due to the different effect of the push-pullsubstituents on the different electronic states, which allows to tune the position of the S2/S1 crossing, opening the way for a wide range of application of such azobenzene derivatives.
[1] Hartley, G. S. The Cis-form of Azobenzene. Nature 140, 281 (1937).
[2] Aleotti, F. et al. Multidimensional Potential Energy Surfaces Resolved at the RASPT2 Level for Accurate Photoinduced Isomerization Dynamics of Azobenzene. J. Chem. Theory Comput. 15, 6813–6823 (2019).
[3] Nenov, A. et al. UV-Light-Induced Vibrational Coherences: The Key to Understand Kasha Rule Violation in trans-Azobenzene. J. Phys. Chem. Lett. 9, 1534–1541 (2018).
[4] Keefer, D., Aleotti, F., Garavelli, M. & Mukamel, S. Imaging conical intersection dynamics during azobenzene photoisomerization by ultrafast X-ray diffraction. Proceedings Nat. Acad. Sci. United States Am. 118, 10 (2021).
[5] Aleotti, F. et al. Spectral Tuning and Photoisomerization Efficiency in Push–Pull Azobenzenes: Designing Principles. J. Phys. Chem. A 124, 9513–9523 (2020).
27 May 2021 – 4pm (UK Time)
Exact-Factorization Based Surface-Hopping for Ultrafast Non-Adiabatic Dynamics
by Dr. Patricia Vindel Zandbergen (Rutgers University)
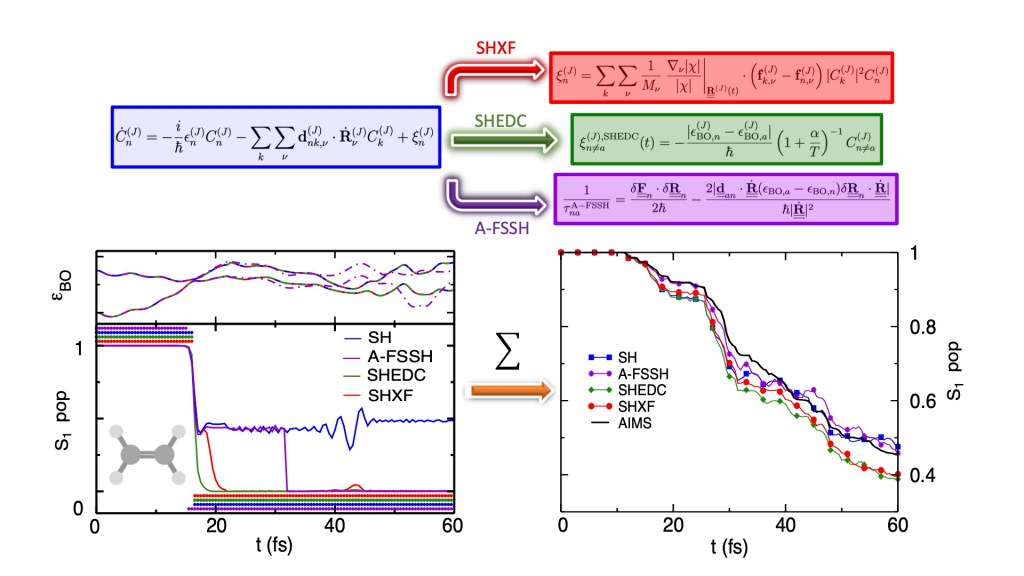
Conical intersections facilitating non-adiabatic transitions have been found to play a key role in many photo-physical pathways. In systems with non-adiabatic couplings between excited states, the correlation between the electron and nuclear dynamics becomes important. Trajectory-based surface hopping dynamics has proven to be a powerful tool to study coupled nuclear-electronic dynamics, but it does not properly account for quantum decoherence. In this work, we present a detailed study of the decoherence correction to surface-hopping that was recently derived from the exact factorization approach. Ab initio multiple spawning calculations are used as a reference for three molecules: ethylene, methaniminium cation, and fulvene, for which non-adiabatic dynamics follows a photo-excitation. A comparison with the Granucci-Persico energy-based decoherence correction, and the augmented fewest-switches surface-hopping scheme shows that the three decoherence-corrected methods operate on individual trajectories in a qualitatively different way, but results averaged over trajectories are similar for these systems.
Reference of this work: https://arxiv.org/pdf/2104.04025.pdf
[1] Abedi, A.; Maitra, N. T.; Gross, E. K. U. Phys. Rev. Lett. 2010, 105, 123002.
[2] Ha, J.-K.; Lee, I. S.; Min, S. K. J. Phys. Chem. Lett. 2018, 9, 1097–1104.
[3] Ibele, L. M.; Curchod, B. F. E. Phys. Chem. Chem. Phys. 2020, 22, 15183–15196.
24 June 2021 – 4pm (UK Time)
Modelling the Ultrafast Electron Attachment Dynamics of Solvated Uracil
by Dr. Cate S Anstöter (Temple University)
Gaining a fundamental understanding of the electron attachment dynamics that lead to DNA damage represents an open challenge to both experimental and computational chemists. Computationally, the challenge arises due to the complex chemical environment, which influences the reactivity of transient species formed. To gain insight into the initial electron attachment dynamics, we employ a bottom-up approach, beginning with bare uracil in the gas phase and expanding to fully solvated uracil. We present a joint DFT molecular mechanics and high-level electronic structure study, modelling the time evolution of both non-valence pre-solvated electron and valence π* electron attached states of solvated uracil. o further elucidate the key physical processes involved in the stabilisation of the different electronic states, we investigate both the effect of the evolving solvent environment and the core geometry of uracil following electron attachment.
