The ISPG just published a series of new articles.

In collaboration with the group of Prof. Petr Slavíček in Prague, we investigated the role of initial conditions when simulating the excited-state dynamics of molecules. This question is particularly relevant if one is interested in the simulation of photoexcitation process triggered by long pulses, i.e., when the formation of a nuclear wavepacket in the excited state is not guaranteed. More information can be found in our article just accepted in Faraday Discussions.
More information in:
J. Suchan, D. Hollas, B. F. E. Curchod, P. Slavíček, On the Importance of Initial Conditions for Excited-State Dynamics, Faraday Discuss., accepted (2018).
The two following papers were published in a special issue of the European Journal of Physics B in honor of Hardy Gross.
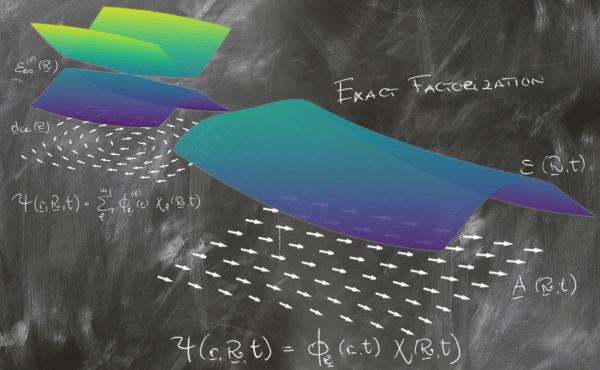
In the first article, we studied with Dr. Federica Agostini the dynamics of a nuclear wavepacket through a conical intersection using the formalism of the Exact Factorization (EF). In a previous work, we looked at a model of the photoisomerization of retinal for this purpose. In this new article, we employed a different model for the potential energy surfaces and played with their diabatic coupling to study how the time-dependent potential energy surface and vector potential – key quantities of the EF – behave in different nonadiabatic regimes.
More information in:
F. Agostini and B. F. E. Curchod, When the Exact Factorization Meets Conical Intersections…, Eur. Phys. J. B, 91, 141 (2018).
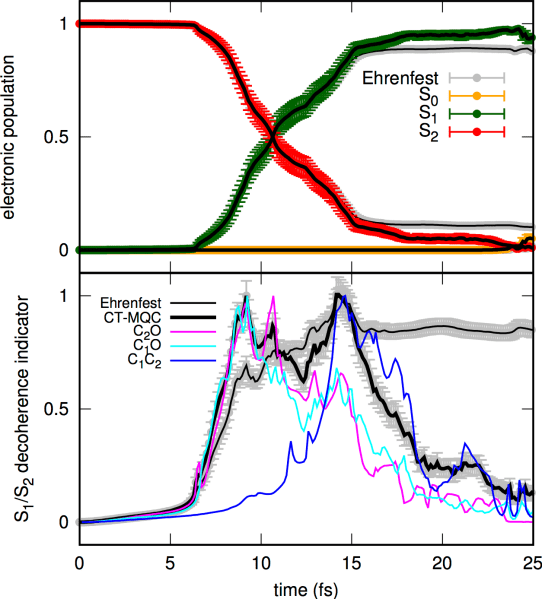
In a second article, we studied the excited-state dynamics of oxirane using the method coined coupled-trajectory mixed quantum/classical (CT-MQC), combined with linear-response time-dependent density functional theory (LR-TDDFT). The results were compared with the Ab Initio Multiple Spawning strategy (also coupled to LR-TDDFT), which reproduces the branching of photoproducts observed in CT-MQC.
More information in:
B. F. E. Curchod, F. Agostini, I. Tavernelli, CT-MQC – A Coupled-Trajectory Mixed Quantum/Classical method including nonadiabatic quantum coherence effects, Eur. Phys. J. B, 91, 168 (2018).









